


In this installment of Expert Picks from DDW 2024, Anisa Shaker, MD, the director of the Swallowing and Esophageal Disorders Center and an associate professor of medicine in the Division of Gastrointestinal and Liver Diseases at the University of Southern California Keck School of Medicine, in Los Angeles, discusses five of her favorite abstracts and presentations from the meeting.
759. The efficacy and safety of guselkumab as maintenance therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 QUASAR Maintenance Study (Rubin et al)
Guselkumab, an interleukin (IL)-23 p19 subunit antagonist, can be an effective and safe maintenance therapy for patients with ulcerative colitis who responded to induction therapy with the drug, according to results of the phase 3 QUASAR maintenance study (ClinicalTrials.gov Identifier: NCT04033445).
Researchers enrolled patients who were responders to IV guselkumab induction in the phase 2b and 3 QUASAR induction studies. These patients all had moderately to severely active UC and previously were intolerant or did not have an adequate response to conventional or advanced therapy.
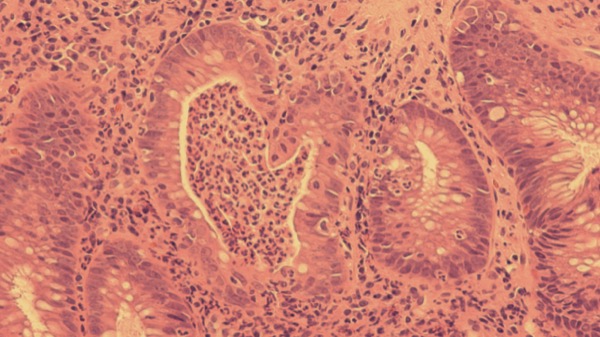
In the maintenance study, patients were randomized 1:1:1 to 200 mg of guselkumab subcutaneously every four weeks, 100 mg of guselkumab subcutaneously every eight weeks or placebo. Outcomes were assessed at week 44 post maintenance initiation.

Of the 568 patients included in analyses, the average age was 40.7 years and average duration of UC was 7.8 years. Nearly two-thirds of patients had severe disease, and more than 40% had prior intolerance or inadequate response to an advanced therapy.
Half of patients who were randomized to 200 mg every four weeks and 45.2% of those randomized to 100 mg every eight weeks achieved clinical remission at week 44. Both of these proportions were significantly higher than the 18.9% seen in patients who were randomized to placebo.
In addition, a significantly higher proportion of patients in each guselkumab arm versus the placebo arm achieved each of the following secondary end points: corticosteroid-free clinical remission, maintenance of clinical remission, clinical response, symptomatic remission, endoscopic improvement, histo-endoscopic mucosal improvement, endoscopic normalization, Inflammatory Bowel Disease Questionnaire remission and fatigue response at week 44.
The researchers also noted that the “safety results through maintenance [week 44] were consistent with the known and favorable safety profile of [guselkumab] in approved indications.”
Dr. Shaker: The results of this trial are quite promising, showing that more patients in the guselkumab maintenance arms than in the placebo arm met the primary end point of clinical remission, as well as multiple secondary end points, including endoscopic and histologic remission. The overall nearly 34% of patients who achieved endoscopic remission with both doses of guselkumab, compared to 15% with placebo, was impressive. Interestingly, the lower and higher maintenance doses of guselkumab showed similar efficacy. This study is encouraging, as it adds to our therapeutic armamentarium for maintenance therapy for UC in patients who have not responded or have been intolerant to prior conventional therapy or advanced therapy with anti–tumor necrosis factor (TNF) agents, vedolizumab (Entyvio, Takeda) or tofacitinib (Xeljanz, Pfizer).
763. Risankizumab versus ustekinumab for the achievement of clinical remission and reduction in inflammatory biomarkers in patients with moderate-to-severe Crohn’s disease: results from the phase 3b SEQUENCE trial (Dubinsky et al)
Anti-TNF–refractory patients with Crohn’s disease taking risankizumab (Skyrizi, AbbVie) were more likely to achieve biologic remission and have early reductions in inflammatory biomarkers than those taking ustekinumab (Stelara, Janssen) in the SEQUENCE trial (NCT04524611).
Building on SEQUENCE results presented at the 2023 UEG Week (abstract LB01) that demonstrated noninferiority of risankizumab versus ustekinumab with respect to week 24 clinical remission and superiority of risankizumab versus ustekinumab with respect to week 48 endoscopic remission, researchers further assessed the timing of clinical remission and changes in inflammatory biomarkers.
Patients with moderate to severe CD who were refractory to anti-TNF therapy were randomized to receive either risankizumab (induction: 600 mg IV at baseline, week 4 and week 8; maintenance: 360 mg subcutaneous every eight weeks starting at week 12) or ustekinumab (induction: single weight-based IV dose at baseline; maintenance: 90 mg subcutaneous every eight weeks starting at week 8).
At weeks 8, 24 and 48, the researchers assessed changes from baseline in high-sensitivity C-reactive protein (hs-CRP) and fecal calprotectin (FCP), as well as the proportion of patients who achieved biologic remission, defined as a Crohn’s Disease Activity Index less than 150 and either hs-CRP no more than 5 mg/L or FCP no more than 250 mg/kg.
By week 8, patients in the risankizumab arm had a greater reduction than those in the ustekinumab arm in both hs-CRP (–10.6 mg/L; 95% CI, –13.0 to –8.2 mg/L vs. –5.5 mg/L; 95% CI, –7.8 to –3.1 mg/L; P<0.01) and FCP (–1,014.0 mg/kg; 95% CI, –1,379.4 to –648.5 mg/kg vs. –650.2 mg/kg; 95% CI –997.8 to –302.7 mg/kg).
Patients taking risankizumab were also more likely than those taking ustekinumab to achieve biologic remission, with the difference being statistically significant by week 24. By week 8, 26.3% of risankizumab versus 21.9% of ustekinumab patients achieved biologic remission; by week 24, 42.8% versus 24.9% patients in each arm achieved biologic remission (P<0.0001), and by week 48, 46.3% versus 27.5% of patients in each arm achieved biologic remission (P<0.0001).
Dr. Shaker: This abstract reports on biomarker data (CRP and FCP) for risankizumab, an IL-23 p19 inhibitor, versus ustekinumab, an IL-12/-23 p40 inhibitor, in patients with moderate to severe CD refractory to anti-TNF therapy. A prior head-to-head trial comparing the efficacy and safety of risankizumab and ustekinumab showed noninferiority of risankizumab for clinical remission at week 24 and superiority of risankizumab for endoscopic remission. In this study, investigators report that reductions in inflammatory biomarkers were greater with risankizumab than with ustekinumab and that this occurred as early as week 8. This study provides additional profiling of another therapeutic option for CD patients.
635. Efficacy and safety of the selective sphingosine 1-phosphate receptor modulator, etrasimod, in adult patients with eosinophilic esophagitis over 52 weeks in the phase 2 VOYAGE study (Dellon et al)
Etrasimod (Velsipity, Pfizer), recently approved for UC, can improve symptoms, histologic findings and endoscopic findings in adults with eosinophilic esophagitis, according to results of the phase 2 VOYAGE study (NCT04682639).
For the first 24 weeks of the VOYAGE study, patients were randomized to either 1 or 2 mg of etrasimod or placebo daily. For the next 28 weeks, patients either continued with their etrasimod regimen or, for the placebo patients, were randomized to switch to either 1 or 2 mg of etrasimod daily.
In total, 30 patients received 2 mg of etrasimod daily for the full 52 weeks, 31 received daily 1 mg of etrasimod for the full 52 weeks, and 24 received placebo for the first 24 weeks and one of the etrasimod regimens for the remaining 28 weeks.
The primary end point of a significant reduction in peak eosinophil count (PEC) at week 16 was achieved in patients in the 2-mg etrasimod arm (placebo-adjusted 46.1% decrease from baseline; P=0.0103). This PEC improvement was maintained at weeks 24 and 52 in those who received 2 mg of etrasimod for the full 52 weeks.
Patients in the 2-mg arm also were significantly more likely to have PEC less than 15 or PEC no more than 6 eosinophils per high-power field and had significant improvements in EoE-Endoscopic Reference Score and EoE-Histology Scoring System grade and stage at weeks 16 and 24 compared with those who received placebo. These improvements also were maintained at week 52 for those who were on the 2-mg etrasimod regimen for the full study period.
Patient Global Impression of Severity and Dysphagia Symptom Questionnaire scores were improved at 52 weeks for etrasimod patients taking either the 1- or 2-mg regimen for the full study period.
None of the 61 patients taking etrasimod for the full 52-week study period experienced serious treatment-emergent adverse events (TEAEs), while three experienced TEAEs that led to treatment discontinuation (including alanine aminotransferase increase, hypoglossal nerve paralysis, dry eye or dyspnea).
Dr. Shaker: This 52-week phase 2 study evaluated the safety and efficacy of the selective sphingosine 1-phosphate receptor modulator etrasimod, which is the first oral small molecule drug under consideration for treatment of EoE. Oral therapy in EoE patients is limited to off-label use of PPIs, topical corticosteroid formulations compounded by patients and/or specialty pharmacies, and a newly approved formulation of the steroid budesonide (Eohilia, Takeda). These therapies may not be effective in nearly 50% of patients. The recently approved biologic dupilumab (Dupixent, Sanofi/Regeneron) is administered subcutaneously and may be ineffective in 40% of patients. So, there certainly is room for improvement in terms of efficacy and patient desirability.
This study showed the safety and tolerability of etrasimod, as well as improvement in several parameters, including histologic and endoscopic scores at 16 and 24 weeks and improvements in clinical symptoms that were sustained at 52 weeks. The study met its primary end point of reduction in PEC at week 16 with the 2-mg etrasimod dose, with 34.6% of patients achieving a PEC lower than 15 at week 52. Overall, this study is sufficiently promising such that it, hopefully, sets the stage for a phase 3 study.
864. Vonoprazan for the treatment of heartburn in non-erosive reflux disease: a randomized trial (Laine et al)
Vonoprazan (Voquezna, Phathom) can quickly and durably alleviate heartburn symptoms in patients with non-erosive reflux disease (NERD), based on results from a U.S.-based clinical trial (NCT05195528).
Investigators randomized adults who experienced heartburn at least four days per week but did not have endoscopic evidence of erosive esophagitis to 10 or 20 mg of vonoprazan or placebo. The patients were followed for 24 weeks, with those initially randomized to placebo re-randomized to 10 or 20 mg of vonoprazan after four weeks. Digital diaries were used twice per day to collect data on heartburn symptoms and use of rescue antacids from patients.
The proportion of days during which patients had no daytime or nighttime heartburn was 44.8% for those taking 10 mg of vonoprazan and 44.4% for those taking 20 mg of vonoprazan, both of which were significantly higher than the 27.7% for those in the placebo group (P<0.0001 for both comparisons).
In addition, the proportion of days without rescue antacid use was significantly higher for those taking 10 mg of vonoprazan (75.9%) and 20 mg of vonoprazan (73.9%) versus placebo (50.0%) (P<0.0001 for both comparisons).
The improvement in symptoms began as early as day 1. Compared with those taking placebo, 8.3% more patients taking 10 mg of vonoprazan and 11.6% more taking 20 mg of vonoprazan experienced no heartburn during the first day of treatment; the differences versus placebo increased to 18.1% and 23.2% for 10 and 20 mg of vonoprazan, respectively, on day 2.
The efficacy was durable, as well, with the treatment effect persisting throughout the 24-week study period.
Dr. Shaker: The potassium-competitive acid blocker (PCAB) vonoprazan has been approved by the FDA for treatment of erosive esophagitis, with a clear role in cases of severe esophagitis (e.g., in scleroderma patients or post-achalasia therapy.) It also was approved previously as part of a formulation for Helicobacter pylori.
This study looks at the role of vonoprazan in patients diagnosed with NERD in the United States. Previous studies in Japan had failed to identify efficacy of vonoprazan for NERD patients. This study showed that both doses of vonoprazan reduced heartburn symptoms quickly—as early as the first day—and that the effect lasted throughout the entire 20-week study period. Why the primary end point wasn’t met in Japan but was met in the United States is not clear, but may be due to differences in CYP2C19 (cytochrome P450 2C19) metabolism.
The quick effect of vonoprazan—as early as the first day—is not surprising, given the pharmacokinetics of PCABs versus PPIs. Where this drug will sit in the therapeutic armamentarium of NERD, a less severe phenotype in terms of complications of Barrett’s esophagus and esophageal adenocarcinoma, has yet to be defined. Long-term safety of such profound acid suppression for NERD also may need consideration. It certainly could be beneficial in up to 10% of patients who continue to have symptomatic, pathologic acid exposure despite twice-daily PPI use.
638. Performance of a machine-learning gene risk score biomarker on predicting response to semaglutide: a prospectively followed multicenter biobank and outcomes registry (Fansa et al)
A machine learning gene risk score (ML-GRS) has the potential to be used to predict response to the glucagon-like peptide-1 receptor agonists semaglutide (Ozempic/Wegovy, Novo Nordisk) and liraglutide (Victoza, Novo Nordisk).
Investigators had developed and reported results for an ML-GRS that can be used to predict response to liraglutide in people with obesity based on the presence of a postprandial satiety (or “hungry gut”) phenotype, which “is associated with greater weight loss in response to GLP-1 receptor agonists” (DDW 2023, abstract 704). In this study, the investigators sought to evaluate whether their model could also be used to predict response to semaglutide.
The investigators had established a biobank and outcomes registry for adults who were receiving weight-loss treatment, and, for this study, they enrolled people with obesity who were taking 0.25 to 2.4 mg of semaglutide.
Based on analysis of saliva or blood, patients were assigned an ML-GRS between 0 and 1. Those with an ML-GRS less than 0.50 were considered to have the hungry gut phenotype. The investigators then assessed total body weight loss (TBWL) every three months for a year and the probability that the ML-GRS would predict semaglutide response (TBWL =5% at one year).
The 84 enrolled patients were an average age of 47.6 years and had an average body mass index of 38.8 kg/m2. The majority (N=51) were hungry gut–positive and female (˜ 80%).
At months 9 and 12, hungry gut–positive patients had significantly greater TBWL than hungry gut–negative patients (nine months: –14.4% vs. –10.3%; P=0.045; 12 months: –19.5% vs. –10.0%; P=0.01). There was no significant difference in TBWL by hungry gut phenotype status at months 3 and 6.
The area under the curve for the ML-GRS for predicting response was 0.76 (95% CI, 0.57-0.94), and the positive predictive value was 0.95.
Dr. Shaker: In addition to being chronic and deadly, obesity is a heterogeneous disease with multiple phenotypes traditionally based on energy balance and behavioral traits. The abnormal postprandial satiety or hungry gut phenotype has been associated with greater weight loss in response to GLP-1s, which prolong postprandial satiety. Ideally, we would be able to determine the factors that predict weight-loss response. The aim of this study was to determine the effect of an ML-GRS in predicting weight loss response to semaglutide, a potentially more effective GLP-1 than liraglutide, for which the model was originally developed.
While this approach requires further study to determine generalizability of results, this study identifies an ML-GRS as a potential novel biomarker that has demonstrated promising results in predicting weight-loss response to GLP-1s. These findings support a novel approach for precision medicine for obesity.
—Compiled and written by Natasha Albaneze, MPH



This article is from the July 2024 print issue.